|
By Sean Marshall
Big news out of the country of India for sickle cell anemia sufferers. According to India.com, India's latest internet news site, it was explained that "scientists in India are developing a portable and affordable diagnostic kit." The article went on to say that "the device could be used by untrained health workers."The people behind the development of the device are from the Indian Institute of Technology-Powai. The idea is to combine the chip that diagnoses sickle cell anemia disease and add it to the modern cell phone. This is all done because the development team feels that many individuals in rural or remote areas cannot afford or even reach medical professionals to have a proper diagnostic. Unfortunately many of these tribal areas have a higher percentage of individuals who suffer from sickle cell anemia disease. So the idea is to allow individuals in "who are not medically trained to be able to diagnoses themselves." According to the article "about five percent of children affected by sickle cell disease die by the age of two." If there are more individuals being able to test for the disease with something as easy to find as a cell phone those numbers could decrees. The device itself will work by adding "having the patient add a drop of their blood to a plastic microfluidic chip that is pre-loaded with the programs required to detect sickle cell disease." The team hopes is that developing countries can use this and save themselves and their children. If you would like to go over the article in more detail the link is provided below: http://zeenews.india.com/news/health/diseases/indian-scientists-craft-portable-diagnostic-kit-for-sickle-cell-anemia_28884.html For those of us who are sports fans interesting news came from twitter last Sunday night. On July 21st Marquise Goodman took to twitter why he left the Buffalo Bills training camp early. He explained that "sickle cell and asthma beat me for today." For those who don't know Marquise Goodman is a wide receiver and kickoff returner for the Buffalo Bills and has had early career success despite his asthma and sickle cell disease. Goodman just finished his rookie year last football season and despite his aliments had a very success year. HIs effectiveness was never questioned despitehaving having sickle cell disease. With a rookie year that involved "17 catches for 238 yards and three touchdowns" his return to the Buffalo Bills was guaranteed. Unfortunately when reports came that he had left camp early Goodman felt he needed to explain himself. Dispite having sickle cell he has been able to have a full rookie year only breaking his hand near the end of the season. Goodman has been known "to want to get out in front of any potential injury talk." Despite being relatively new to the sports world he is a beacon to those who suffered from any illness brought on by sickle cell that the disease doesn’t stop you from being active or even pursuing dreams of being a professional athlete. If you would like to know about the article head over to the link: http://bills.buffalonews.com/2014/07/21/marquise-goodwin-wants-world-know-hes-hurt/ If you have any questions or concerns email us at [email protected]
0 Comments
By Sean Marshall
Important news for parents of children suffering from sickle cell anemia, according to Health-e-galaxy, a global health news and information website, "up to 41% of children with sickle cell anemia may have some degree of obstructive sleep apnea syndrome." This came from a Sleep and Asthma Cohort study out of New York State. According to the study children who have sickle cell anemia are more likely to have obstructive sleep apnea syndrome or OSAS for short. Normally OSAS affects 1% to 5% of children which is not very high. However when children with sickle cell anemia disease were tested it was much higher. Tests showed that the sickle cell children had a higher rate of OSAS. "Among 243 children in the final sample, 41% had an obstructive apnea hypopnea index (OAHI) of at least 1, 34% had an OAHI of at least 1 plus habitual snoring, 24% had an OAHI of at least 2, and 10% had an OAHI of 5 or greater." Sleep apnea causes "a person to often awaken with shortness of breath or have a difficult time getting to sleep or staying asleep. Like with obstructive sleep apnea, snoring and daytime sleepiness can occur." This means that less air is getting that there is a lack of oxygen getting to the body; this is especially problematic for those suffering from sickle cell anemia because it just adds more complications for the human body. The article stressed the importance of testing early to avoid complications later on. Measured could be taken such as surgeries or sleep therapy to aid in proper sleep. If you would like to learn more follow the link: http://www.hegalaxy.com/obstructive-sleep-apnea-common-in-kids-with-sickle-cell-anemia/ Good news for the Democratic Republic of Congo. The Gertler Family Foundation according to a press release in the digitaljournal.com "saving the lives of hundreds of these children and reducing infant mortality in the Democratic Republic of the Congo." The press release outlined that The Gertler Family Foundation or GFF had combined their efforts with Dr. Baron Ngasia, one of the founders of the Sickle Cell Disease Research Network in Central Africa and have "established a SCD unit at Lumbu Hospital in Kindu, Maniema Province, where he is also the medical director." The GFF plans on "continuing research, screening and treatment management for newborns with SCD, as well as educating parents about SCD and training medical staff about the disease." It was then stated that in May 2014 at GFF Symposium held in Kinshasa, the Democratic Republic of Congo's capital. At the symposium "more than 150 participants from across the DRC, Africa, Europe, and the United States gathered to discuss establishing a national SCD program in the DRC." It was explained that "all parties would be working with the Ministry of Health to develop SCD policy; and improving the knowledge and SCD practical skills of health care providers." GFF's end goals were to combine early screen for newborns with "specific immunization and penicillin prophylaxis to allow for better management of the disease in life." Other goals included management of the disease in DRC and other African countries. Despite the overwhelming good this organization is doing the news must be taken with a grain of salt after all this is a press release and that means its company propaganda as much as it is good news. If you would like to see the Press Release for yourself just head to the link: http://www.digitaljournal.com/pr/2057491 If you have any comments questions or concerns email us at: [email protected] By Sean Marshall
Good news for those who are suffering from sickle cell anima or just have any questions about the disease itself. According to Huewire.com, an online source for news in Mississippi, a treatment center for Sickle cell disease has just opened up in Missouri. The center's opening took place on the 17th of June and is located at the University of Mississippi Medical Center. Unfortunately many media sources had not picked up on such an important milestone for those suffering from sickle cell anemia. The good news however is that the clinic is "meant to give specialized care to realize the chronic excruciating pain sufferers of this inherited disease must go through." What makes the clinic so important is that it is one of the first in its kind. Before its opening any individual suffering from sickle cell anemia complications would "have to rely on emergency rooms where waiting times could range from minutes to hours." This put a lot of people at risk but now with the center managing the flow of patients not just the people with sickle cell disease but clearing up emergency rooms as well. The article did go on to say that "anyone suffering from sickle cell anemia is still more then welcome to UMMC emergency rooms they are still more then welcome there and can still receive treatment If you would like to know more follow the link below: http://huewire.com/health/sickle-cell-anemia-treatment-clinic-opens-mississippi/2204/ Other sickle cell anima news from the states involves the NFL twins Devin and Jason McCourty raising sickle cell awareness. Devin, a 2010 first-round pick of the Patriots and twin brother Jason, who plays for the Titans have partnered with Embrace Kids foundation. The twins have lost close family to the disease and according to thepostgame.com plan on spreading awareness of this crippling disease. It was explained in detail that "Their father carried the trait, so the twins were tested to see if they had it as well. Fortunately for them, they do not. But after their father passed, the two grew close to their aunt and uncle who do have the disease." The most exciting news was that the twins plan on using their fame as NFL players to launch a "Public Service Announcement (PSA) to kick-start Sickle Cell Awareness Month." The two seem to have to have further plans involving raising more money for research and even more awareness. If you would like to learn more about the NFL twins story or how you can help follow the link: http://www.thepostgame.com/blog/more-family-fun/201407/devin-jason-mccourty-nfl-rutgers-sickle-cell-embrace-kids Lastly in medical breakthrough news another discovery towards ending sickle cell animea has been made. According to healthcanal.com's latest article concerning sickle cell animeia, "A compound discovered at Virginia Commonwealth University has taken yet another step closer to becoming the only approved drug in the world that is therapeutically effective in managing adult sickle cell disease by targeting hemoglobin." The purpose of the Aes-103 is to "bind to and stabilizing the relaxed state of hemoglobin and increasing oxygen affinity and stabilization, thereby reducing the sickling of red blood cells which, in turn, may reduce sickling-related outcomes such as vaso-occlusive crisis, pain, severe anemia and fatigue." According to the report the medical compound is called Aes-103 the compound is one of the first steps in medical breakthrough for pain tolerance brought on by hemoglobin imbalances. The Aes-103 drug production program is "currently in a Phase 2 clinical trial as part of an ongoing collaboration with the NIH’s National Center for Advancing Translational Sciences." This means the drug is on its ways to human testing in no time. If you would like to learn more about Aes-103 and other early stage medical drug testing go to the link below. http://www.healthcanal.com/medical-breakthroughs/52894-another-step-toward-sickle-cell-disease-treatment.html If there are any comments questions or concerns about what has been said here feel free to send an email to [email protected] 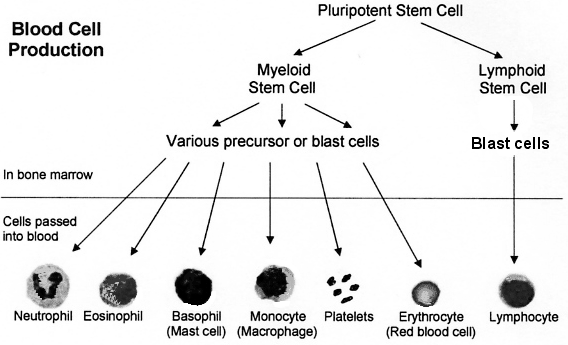 ~The Most Curable Option for SCD~
Author: Kara Martina, B. Sc. Biopsychology More than often we hear news stories about this elusive Bone Marrow Treatment and how it helps those with Sickle Cell Anemia. Despite its success, many people remain ignorant about Bone Marrow Transplants (BMT) and its potential life-saving effects for those with anemia or cancer. This lack of knowledge leaves people apathetic and unaware of how to become a Bone Marrow donor. So: what are Bone Marrow Transplants and how do they work? Are they Successful? According to Blood Journal, a matched Bone Marrow transplant remains the only curative strategy for Sickle Cell Diseases (1). The first success was reported in 1984 in a patient with coexisting acute myeloid leukemia (2). Not without potential grafting complications and extensive conditioning regimens, Bone Marrow Transplants are considered effective. The greatest success rate was reported in 2011 with overall survival and event-free survival of 93% and 86%, respectively (3).The most recent article published in JAMA, July 2 2014 announced that 29 out of 30 Sickle Cell patients successfully underwent a matched Bone Marrow Transplant (4). How does it work? Bone Marrow donors are matched based on immune system markers, and once an allogeneic match is found, their bone marrow is harvested. More likely to be a brother or sister, a World Wide Marrow Bank is also used to find close matches. The replacement bone marrow cells grow into normal blood cells without the sickle-shaped deformities. What is Bone Marrow? Where your body makes the components of blood. Bone marrow is the soft sponge-like material in the centre of bones. Large flat bones such as the breastbone (sternum) and pelvis contain the most bone marrow (5). To make blood cells constantly you need a healthy bone marrow. What does Bone Marrow in Sickle Cell Patients look like? From the beginning, stem cells from the bone marrow in a person with Sickle Cell Anemia are programmed to produce Sickle Blood cells. How are BMTs done? First, the defective bone marrow needs to be partially or fully wiped out. A course of radiation or chemo or both is used to wipe out defective marrow. Often the hardest and most tiring time during BMT, this first step alone has taken lives before the procedure can even be completed. Difficult for the individual and the family, this chemo/radiation takes a heavy toll on the body and often perturbs people form choosing BMT. More research into creating less devastating effects of the first stage continues. Second, the healthy bone marrow is injected, delivered into the patient’s bloodstream (6). Described by some as ‘similar to getting a blood transfusion,’ the stem cells travel through the blood into the bone marrow. Most times, no surgery is needed. How can I donate/become a bone marrow donor? Simply register! To become a donor it only takes a small vial of blood or swab of cheek cells to be typed as a bone marrow/stem cell donor. It’s like saying to anemia and cancer communities alike, “I’m here!” Minor costs may apply for HLA tissue typing. If someone needs your bone marrow, you’ll be contacted and harvested. Screening is often rigorous, but given the benefits of healthy bone marrow—it’s worth the effort! Individuals wishing to register can: · Call 1-888-236-6283 (1-888-2-DONATE) · Create an account and appointment with blood.ca (create username and password) · Contact Kyshah Powell at: [email protected] · Outside of North America visit here. References 1. M.M. Hsieh, C.D. Fitzhugh and J.F. Tisdale. Allogeneic hematopoietic stem cell transplantation for sickle cell disease: The time is now. Blood. Vol. 118, August 4, 2011, p. 1197. doi: 10.1182/blood-2011-01-332510. 2. Johnson FL, Look AT, Gockerman J, Ruggiero MR, Dalla-Pozza L, Billings FT 3rd. Bone-marrow transplantation in a patient with sickle-cell anemia. N Engl J Med 1984;311(12):780-783. 3. Bernaudin F, Socie G, Kuentz M, et al. Long-term results of related, myeloablative stem cell transplantation to cure sickle cell disease. Blood 2007;110(7):2749-2756. 4. M. M. Hsieh et al. Nonmyeloablative HLA-Matched Sibling Allogeneic Hematopoietic Stem Cell Transplantation for Severe Sickle Cell Phenotype. JAMA. Vol. 312, July 1, 2014, p. 48. doi:10.1001/jama.2014.7192. 5. Kenny, Tim. (2012). Stem Cell Transplant. Retrieved July 10, 2014 from http://www.patient.co.uk/health/Stem-Cell-Transplant.htm 6. A. D. A. M. (2013). Bone Marrow Transplant. The New York Times. Retrieved from http://www.nytimes.com/health/guides/surgery/bone-marrow-transplant/overview.html By Sean Marshall
Big announcements in Sickle Cell treatment this week. The first bit of good news comes from CTVnews.ca where it was discovered that an American study showed that bone marrow transplants reversed sickle cell disease in some adult patients. According to the article "bone marrow transplants reversed sickle cell disease in 26 out of 30 adults." Even better news the study also discovered that "15 of the patients were able to stop taking drugs that prevent rejection of the procedure just one year later." According to the author of the article, Lindsey Tanner, the bone marrow treatment involves taking bone marrow from a brother or sister the idea is that "the brother or sister whom have stem cell-rich bone marrow and is a good match for the patient." The process also involves chemotherapy and radiation treatment to break down the bone marrow of the individual with sickle cell so it can then be successfully replaced with the donor tissue. However the treatment isn't foolproof. There is still research and testing to be done for this to be the cure for sickle cell disease but for the individuals who were lucky enough for it to work their future is looking even brighter. If you would like to see the information yourself here's the link: http://www.ctvnews.ca/health/bone-marrow-transplants-reverse-sickle-cell-disease-in-some-adults-u-s-study-1.1894816 Or if you would like to read a similar story go here: http://www.newsmax.com/scitech/sickle-cell-reversed-transplant/2014/07/01/id/580333/ Unfortunately all news cant be good news. A news article out of AllAfrica.com reports that seven million Ugandans risk producing offspring with sickle cell disease. According to the report "studies by the Ugandan Sickle Cell Association show that Bamba people in West Uganda carry 45% of the sickle cell gene." If the numbers are accurate and up to date tat means that the carry the highest rate in the world. The article went on to explain that with better medical equipment those suffering could be treated and even diagnosed early to avoid unnecessary complications. Link to article: http://allafrica.com/stories/201406300939.html Other good news about potential sickle cell treatment comes from DigitalJournal.com. According to their July second article the latest drug designed to treat pain caused by sickle cell disease has passed its clinical trial. It was explained that the research team at the US Davis School of Medicine was conducting clinical trials on sickle cell treatment drugs. One drug that passed the clinical tests is called GMI 1070 and has major positive affects. It is designed to "maintain adequate blood concentrations in sickle cell victims." According to the report from The Digital Journal the drug when they spoke to Ted Wun the associate dean for research at the US Davis School of Medicine, he explained that the drug "did improve blood flow and reduce the markers of cell activation." GMI 1070's purpose is to reduce the chance of red blood cells from sticking to white blood cells and one another. This is what causes the damage to the body of those who suffer from sickle cell disease. The article did point out that the whole purpose of the trials was to prove that the drug is safe for human testing and according to the article it is. The next step is human testing but if the trials are as positive as they were in animal testing this could be a big breakthrough in sickle cell disease management. If you would like to learn more about GM 1070 follow the link provided: http://www.digitaljournal.com/pr/2032018 If you have any questions or concerns just email us at: [email protected] ~Iron Overload~ Author: Kara Martina, B. Sc Biopsychology Sickle Cell Patients often need blood transfusions as their own blood inadequately meets the body’s needs. These transfusions decrease early blood cell death, restrict endothelial damage and increase the blood’s oxygen carrying capacity. Transfusions for SCD patients prevent strokes and treat acute chest syndrome. Routinely administered and prescribed, blood transfusions deal with a range of additional SCD complications, such as priapism, vaso-occlusive crises, leg ulcers, pulmonary hypertension, and during complicated pregnancies (1). While transfusions have countless success stories, unfortunately a toxic side effect accompanies it: Iron overload.  Iron overload, the unavoidable complication of transfusions, leads researchers to hope for alternatives to this defaulted treatment. Although the relation between SCD patients and Iron overload needs further research, it has been suggested that Iron overload for those with SCD should not be underestimated (2), especially in those with chronic transfusions (3). With repeated transfusions, proteins in your plasma called serum transferrins become saturated with iron. The unwanted Iron, now called Non-Transferrin-Bound Serum Iron (NTBI) enters cells with high redox potential causing unwanted reactions which can eventually lead to organ damage (4). In other words, excess Iron causes excess free radicals that may kill your cells. For those with ß-thalassemia major, Iron overload can be lethal. For those with SCD, it should not be ignored. Symptoms range from tiredness and weight loss to heart failure and liver disease (5). To combat iron overload, drug options include Deferiprone, Deferoxamine, and Deferasirox. Proven to prevent organ failure and other complications, all three come with their prospective side effects. Deferiprone: Binds to Iron radicals. Commonly given in 500mg tablets. Side effects: Nausea, Chromaturia, and Abdominal Pain. May cause lethal agranulocytosis. Deferoxamine: Forms a stable complex with Iron. Also commonly given in 500mg tablets. Should not be taken with Vitamin C tablets . Side effects: Injections site reactions, abdominal pain, muscle spasms. Deferasirox: Reduces liver iron concentration and serum ferritin levels. Commonly given in either 125mg to 500mg tablets. Side effects: Serum creatinine increase, Abdominal pain, diarrhea, rash. May cause acute renal failure and death. All information was retrieved from the credible MedScape App available at http://itunes.apple.com or the App Store on your smart phone (6). Given the amount of drugs on the market and variability of each anemia case, this free App helps patients learn more about the drugs prescribed to them. Blood transfusions treat the side effects of SCD but further research on discovering less harmful iron chelation drugs would benefit the SCD community and anyone suffering from anemia.
References 1. Radha Raghupathy, Deepa Manwani, and Jane A. Little, “Iron Overload in Sickle Cell Disease,” Advances in Hematology, vol. 2010, Article ID 272940, 9 pages, 2010. doi:10.1155/2010/272940 2. E. B. Fung, P. Harmatz, M. Milet, et al., “Morbidity and mortality in chronically transfused subjects with Thalassemia and Sickle Cell Disease: a report from the multi-center study of iron overload,” American Journal of Hematology, vol. 82, no. 4, pp. 255–265, 2007. 3.Paul Harmatz, Ellen Butensky, Keith Quirolo, Roger Williams,Linda Ferrell, Thomas Moyer, Daniel Golden, Lynne Neumayr, Elliott Vichinsky., “Severity of iron overload in patients with sickle cell disease receiving chronic red blood cell transfusion therapy,” Blood Journal, 2000,96(1)76-79. 4. W. Breuer , Z. I. Cabantchik, B. P. Esposito, C. Hershko, P. Pootrakul and P. Sirankapracha, “Labile plasma iron in iron overload: redox activity and susceptibility to chelation,” Blood, vol. 102, no. 7, pp. 2670–2677, 2003. 5. Aplastic Anemia MDS International Foundation. (2011). Iron Chelation Therapy. Retrieved July 4, 2014 from http://www.aamds.org/node/82 6. WebMD Health Corporation (2012). Medscape (Version 4.0) [Mobile application Software]. Retrieved from http://itunes.apple.com |
AboutThis section is solely to let our Sickle Soldiers tell their story trials & tribulations alongside things they feel are wrong in the Sickle Cell Community Archives
March 2016
Categories
All
|
 RSS Feed
RSS Feed
