|
By Sean Marshall
Another step forward this week for drug treatment of sickle cell anemia. According to jamanetwork.com, JAMA standing for The Journal of the American Medical Association, there is a new sickle cell drug candidate under development. In a previous news update from thesickleinme.com we mentioned a new drug called Aes-103 and how it was being tested on animals in the hope to one day fight sickle cell disease in human beings. The good news is that The Journal of the American Medical Association has reported that for the first time “a pharmaceutical company has acquired a drug candidate developed with resources from a National Institutes of Health program for rare and neglected diseases.” The top disease on the list is in fact sickle cell anemia. According to JAMA “The drug, called Aes-103, treats sickle cell disease by binding directly to hemoglobin and changing its structure.” In other words the drug reduces “the sickling of red blood cells.” Not only can it reduce the sickling of red blood cells but “it is the first drug specifically developed to target the underlying molecular mechanism of sickle cell disease.” If you would like to learn more about the drug you can go to JAMA’s website and read the article here: http://jama.jamanetwork.com/article.aspx?articleid=1899183 New scientific findings link sickle cell and malaria. In an update from sciencebriefs.com, The Duke Medical Center have discovered a link between the micro RNA, RNA being an even smaller biological marker the DNA, in sickle cells and growth of the parasitic activity that causes malaria. Duke University originally set out to test genetic material in red blood cells to figure out what could slow down malaria growth. It was explained that malaria is caused by “a parasitic gene regulation,” this gene regulation allows the virus to cause sickness. The article mentioned that “Duke Researchers discovered that genetic material in red blood cells may help alter parasite activity via a novel mechanism that changes parasite gene regulation.” Acceding to a quote from Dr. Jen-Tsan Chi a senior author and associate professor “sickle red cells directly participate in the gene regulation of malaria parasites,” Dr. Jen-Tsan further explained that “These microRNAs enriched in the sickle red cells reduce the parasite’s ability to propagate, so that certain people stay more protected.” With this scientific advancement, in an ironic twist one day sickle cell disease may just save lives. If you would like to learn more about malaria and its links to sickle cell you can follow the link provided: http://www.charlotteobserver.com/2014/08/24/5118987/science-briefs-sickle-cells-combat.html#.U__ggKNTxtN Or if you would like to read the original journal of medicine research you can go to: http://www.corporate.dukemedicine.org If you have any comments questions or concerns email us at [email protected]
0 Comments
~A Discussion on Bone Marrow Transplants~ Author: Kara Marina B. Sc. Biopsychology Located in your bone marrow, Hematopoietic stem cells eventually make the components of blood. Similar to embryonic stem cells, hematopoietic stem cells are categorized into four types: pluripotent, totipotent, multipotent or unipotent (1). Their potency depends on how far in their development they exist, whether they can become anything or just one type of cell. Unlike embryonic stem cells, hematopoietic cells can only differentiate into blood cells, but still the possibilities are vast. 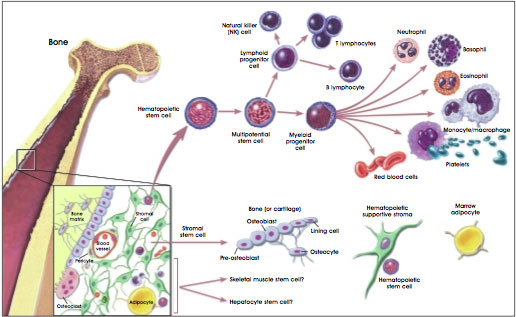
Sickle Cell patients require red blood cells be produced from their Bone Marrow Transplant (BMT). Leukemia patients require all of their blood components be replenished, each blood cancer arresting bone marrow cells at different stages. The best stem cell has a wide range of development possibilities: the pluripotent and the totipotent cells. Also, the best stem cells can regenerate themselves after they die. These long-term stem cells are sought after and hoped for while harvesting. Unfortunately, to date, there is no method to discern the difference between stem cells with longevity and cells dubbed short term-progenitor cells (2). These short term-progenitor cells may last four to five months.
Two theories surround the differentiation of bone marrow cells: the deterministic view and the stochastic view. The deterministic view contents that each cell becomes a product of its environment, changing in response to external signals such as hormones. The stochastic view contents that stem cells differentiate randomly. Is it random or is it fate? Research shows us both: random and fate. Totipotent Hematopoietic cells differentiate deterministically and therefore have the greatest ability to react appropriately to a new host’s needs. But each particular cell’s fate seems to be random within that environment even though the overall effect is determined by the host. In the end, there’s an order to their random, as seen over a long period of time. But only true stem cells can show us this glory: pluipotent or totipotent stem cells capable of self-renewal. Retrieving these cells makes the difference between a successful and a non-successful BMT, among other factors, namely having a great match! Fortunately, new ways of extracting stem cells are quickly replacing the old bone marrow needle routine. Because a small amount of stem cells circulate in the peripheral blood, researchers have found they can ‘coax’ the bone marrow cells from the bone marrow into the circulating blood by injecting a cytokine such as granulocyte-colony stimulating factor (3). Injected prior to harvesting, this procedure yields more Hematopoietic cells with better results! All the more reason to register….. Individuals wishing to register can: · call 1-888-236-6283 (1-888-2-DONATE) and ask about registering to be a bone marrow donor · create an account and with blood.ca (create username and password) and receive a mouth-swab kit by mail · Contact Kyshah Powell at: [email protected] for the September drive in Hamilton · Outside of Canada visit here. References 1. Murnagham, Ian. (2014). Stem Cells: The Facts. Explore Stem Cells. Retrieved August 27, 2014 from http://www.explorestemcells.co.uk/StemCellsFacts.html 2. Percorino, Lauren. (2012). Stem Cells for Cell-Based Therapies. Actionbioscience. Retrieved August 27, 2014 from http://www.actionbioscience.org/biotechnology/stem_cells_for_cell-based_therapies_article_update.html 3. National Institute of Health. (2011). Hematopoietic Stem Cells. In U.S. Department of Health & Human Services. Retrieved August 27, 2014 from http://stemcells.nih.gov/info/scireport/pages/chapter5.aspx#figure1 By Sean Marshall
Bizarre findings for adolescents suffering from sickle cell anemia, according to Healio.com adolescents and young adults with sickle cell disease are statistically more likely to have chlamydia. The information comes from an article on Healio.com. The website is generally known for being a web site dedicated to spreading knowledge of infectious disease in children and young adults. The information comes from a study conducted by Tulane University School of Medicine in Mew Orleans. The testing assessed "the burden of asymptomatic chlamydia among patients with sickle cell disease." According to the results of the testing 12% of participants were tested positive for chlamydia and sickle cell disease. The interesting aspect was that “adolescents made up three-quarters of positive results.” If you would like to learn more about the study follow the link: http://www.healio.com/pediatrics/emerging-diseases/news/online/%7B8fe91aab-4d16-4807-9027-b83e1e83fd52%7D/adolescents-young-adults-with-sickle-cell-disease-have-higher-rates-of-chlamydia?sc_trk=internalsearch Other interesting developments this week include a study that reveled patients with sickle cell anemia have lower levels of vitamin D. On 7thspace.com a study was revealed that “ the average vitamin D levels are also substantially lower in African Americans than whites,” it also went on to say “that the population distributions vitamin D among Jamaicans of African descent and West Africans are the same as among USA whites.” It was explained that these results were found by collecting samples from Jamaican and West Africans who suffer from sickle cell disease have a 37% and 39% higher chance of having a lower level of vitamin D in their system. The reason why the study was conducted is that this genetic anomaly is happening across the world to sickle cell sufferers. The team responsible for the study feel that “this issue deserve further investigation, and a randomized trial is warranted to address efficacy of supplementation on sickle cell anemia.” If you’re interested in more details about the study go to: http://7thspace.com/headlines/480269/vitamin_d_levels_are_low_in_adult_patients_with_sickle_cell_disease_in_jamaica_and_west_africa.html Good news for children who suffer from sickle cell disease. A new study from the New England Journal of Medicine has explained that monthly blood transfusions may lower the chance of silent strokes in children with sickle cell. NursingKnowledge.com posted an article on their site outlining all the details. The study was conducted by Dr. James Casella who first started out by using MRI technology to scan over 1,000 brains of children between the ages of 5 to 15 with sickle cell anemia. The team was looking for past silent strokes and other similar brain activities. The study found that there were 196 children who had had silent strokes. The 196 children were then separated into two groups. One group was given monthly blood transfusions while the other was given regular care. In the three year study those children who had been given regular blood transfusions were less likely to have silent and full blown strokes. It was explained that this process did not fully remove the chance of strokes but did decreases it. There were however some complications the article pointed out that “the kids have to be monitored for side effects like iron overload, which is very common.” It was also explained that “excess iron in the blood is potentially dangerous because the mineral can damage organs, and it may require treatment with special drugs that draw excess iron from the blood. Steinberg did feel that the study was not long enough and that there needs to be more testing for both long and short term side effects. If you would like to learn more about the study head to: http://www.nursingknowledge.org/nursing-news/blood-transfusions-may-cut-risk-of-silent-stroke-in-kids-with-sickle-cell or head to the link below for another view on the issue: http://www.eurekalert.org/pub_releases/2014-08/nion-mbt081814.php If you have any questions or concerns email us at [email protected] By Sean Marshall
This week there was another update on the treatment of sickle cell anemia pain with medical marijuana. According to ABC’s channel 7 news vaporized medical marijuana is getting the green light for testing on human subjects. Originally the study almost didn’t happen. Many states do not approve of the use of medical marijuana as a pain suppressor. This meant that all testing had to move to San Francisco, where it is legal to use medical marijuana. Even then the study still had to go through vigorous animal testing phases before even allowing the study to begin on humans. According to the report the reason why marijuana is even being considered is because many of the breeds of marijuana being used in the study contain a chemical called CBD. Doctor Abrams, one of the individuals working on the testing, explained why CBD is so important "CBD is another cannabinoid, one of the active ingredients in the plant. It's not psychoactive, but it is active against inflammation and pain.” This initial discovery was made when mice that were genetically programmed to develop sickle cell disease were given certain pain reducing drugs. The mice that were treated with marijuana that was high in CBD showed “less pain and less inflammation.” The real challenge, according to the article was providing a way to get the CBD into the brain in the least harmful way possible. The way the team handled this was vaporizing the plant. According to team members “Vaporizers provide one of the safest ways to ingest the medicine because you're not actually burning any plant matter.” Luckily after a yearlong study by the FDA had found that “vaporized CBD was not harmful to mice or dogs,” the testing can now start on human subjects. There are some ethical issues involved such as genetically modifying animals to have sickle cell disease, or the use of medical marijuana but the article only addresses the medical issues of pain treatment. All in all any step forward in sickle cell treatment is usually a good one. If you would like to learn more about the process involving vaporization you can go to the link: http://abc7news.com/health/vaporized-medical-marijuana-study-given-green-light/261437/ Other news this week involved an article from News Works an online source of news in the Philly region. The article is actually filled with all sorts of interesting sickle cell stories and points of view from the day in the life of a young boy with sickle cell disease to section on disparities and diagnosis. The particular section that seemed so unique was called “The Forgotten Disease.” In this part of the article it addressed the issues the public has with sickle cell. The section deals with the opinions of Doctor Kwaku Ohene-Frempong and like the title indicates it is he feels sickle cell disease has been forgotten. He explains that "People used to hear a lot more about sickle cell disease in the 70s.” He goes on to say that because of all the inactivity some of the population could think that the disease has been cured. It is explained by News Works that sickle cell disease has been documented and studied for over 100 years but there is still no known cure. It is mentioned that a tone point that the medical community felt that once gene therapy was developed there could be a way to cure sickle cell disease. Yet Ohene-Frempong explained that “we've had gene therapy for 15 years or so and not a single bona fide attempt at gene therapy has been done on sickle cell disease." Despite life expectance and drug therapy for pain have increased no real attempts at gene therapy related cures have been made. This is why Ohene-Frempong feels that sickle cell is the forgotten disease. If you would like to view the entire article and read up on other sickle cell issues in the Philly region follow the link provided: http://www.newsworks.org/index.php/local/item/71459-sickle-cell-disease-still-persists-100-years-after-discovery?linktype=hp_topstory If you have any questions, comments, concerns or even ideas for future projects send an email to: [email protected] Reply Forward By Sean Marshall
Good news from The Washington Post, according to an article published on washingtonpost.com doctor Griffin Rodgers along with The National Institute of Health or the NIH for short. His team according to The Washington Post has created “the first effective therapy for sickle cell disease.” The NIH researchers along with Rodgers have now developed a “modified blood stem-cell transplant regimen.” The regiment itself is highly effective in reversing sickle cell in adults. According to the article these findings were based off of a clinical trial involving 30 adult patients. If you would like to learn more about Rodgers or the article follow the link: http://www.washingtonpost.com/politics/federal_government/nih-scientist-transforming-treatment-of-sickle-cell-disease/2014/08/05/bafe3d4c-1ca6-11e4-ae54-0cfe1f974f8a_story.html Shocking news out of Nigeria this week, at least 100, 000 babies die from sickle cell disorder every year according to statistics gathered by the World Health Organization. These statistics were first brought to the public’s attention by PM news, one of Nigeria’s news organizations. If PM News is right the world Health Organization (WHO) “Nigeria showed 75 per cent of infant sickle cell cases in Africa and almost 80 per cent of infants die from the disease in the continent.” It is also explained that Nigeria has the largest population of individuals affected by sickle cell disease. The article gave further evidence of this fact including such startling numbers like “over 40 million Nigerians as carriers and an estimated 1,000,000 living with sickle cell disorder in Nigeria.” Because of the staggering numbers many efforts have been out in place including the Tolulope Akinduro Foundation, a center for hemoglobin disorder management and prevention center. The center was named after a young doctor who passed away July 15 2013 from sickle cell anemia. According to the National Director of SCFN, Dr. Annette explained the easiest way to combat sickle cell anemia disorder is with genetic counselling. “Genetic counselling is giving unbiased information in a structured cohesive manner. It is a gateway for effective care and management of sickle cell anemia.” If you would like to read more about Dr. Annette, Tolulope Akinduro or just more information about the article go to the link: http://www.pmnewsnigeria.com/2014/08/06/100000-babies-die-of-sickle-cell-disorder-in-nigeria-annually-who/ A United States court case involving sickle cell anemia went public this week. The news appeared on courthousenews.com and covered the death of a Berkly football player with sickle cell anemia. In Oakland California a university football player by the name of Erik Plancher died due to an over intensive work out his trainer. According to the report Plancher’s family felt the training drills were “extremely intensive and egregiously inappropriate, given his condition.” The defendants in this case being is the regents of the University of Central Florida. However the case itself had named the head athletic trainer at the time a one Robert Jackson. The Plancher family was awarded “10 million dollars against the UCF Athletics Association.” No further details have surfaced. If you would like to learn more about the case head to the link provided: http://www.courthousenews.com/2014/08/07/70179.htm If you have any questions or concerns email us at [email protected] ~Sickle Cell Kidneys: Complications, Treatment and Awareness~ Author: Kara Martina, B. Sc. Biopsychology Those with Sickle Cell Disease and Trait have a different physiology than the general population. Having to be keenly aware of unique problems, this article aims to cover the common Sickle Cell complications and lingo of the kidney: Sickle Cell Nephrology. Routine Doctor check-ups prevent long-term damage and catch kidney problems before they escalate. Responsible for filtering the waste products from the blood, the kidneys need more than a basal level of functioning to keep the human body healthy. When they don’t work properly, the ramifications can be detrimental or life changing, with treatment ranging from transfusions to dialysis. Those with SCD or SCT fall under a category of at-risk individuals for kidney diseases. Nephrology knowledge should be heeded and spread: Sickle Cell Nephrology: A Summary Glomerulonephritis
Symptoms: Often asymptomatic, but Nephrotic and Nephritoc symptoms can occur: proteinuria, hematuria. Treatments: Transfusions, Angiotensin-converting enzyme (ACE) inhibitor medicines. Hematuria: Blood in the urine.
Symptoms: Pink, reddish urine, painless. Treatment: Bed rest and maintenance of high urine flow rate, iron replacement and/or blood transfusions. Hemosiderosis: Iron overload in the kidneys. Symptoms: Joint pain, fatigue, unexplained weight loss, abdominal pain, abnormal bronze or gray skin color. Treatment: Limiting blood transfusions and starting iron chelating therapy. See your doctor about chelating drugs. Microalbuminuria/proteinuria: An increase in urinary excretion of the protein albumin. Symptoms: Sometimes asymptomatic. Detected by measuring aluminum-creatinine ratio (ACR) in a spot urine sample where 30-300mg of albumin will be detected. May cause urine to look foamy, swelling of the hands and feet; edema. Treatment: angiotension-converting enzyme (ACE) inhibitors or angiotension receptor blockers (ARBs). Nocturia: low bladder capacity and production of a large volume of urine when you sleep. Symptoms: Having to wake up during the night to urinate. Treatment: See your physician. Treatments will include interventions such as restricting fluid in the evening, elevating legs, wearing compression stockings. Medications include: Desmopressin (DDAVP), Bumetanide (Bumex), Furosemide (Lasix), Anticholinergic drugs. Papillary Necrosis: Part or all of the kidney’s papillae die. The renal papillae are the areas where the collecting ducts meet the kidney and the urine flows into the ureters. Symptoms: Back pain, flank pain, bloody urine, cloudy urine, dark urine, tissues in the urine, chills, fever. Treatment: Exchange transfusions, replacing losses, maintaining hydration, alkalinizing the urine. Polyuria: Large production of urine. Symptoms: Increase in urinary frequency, greater than 3 L/day. Treatment: Admission to hospital, fluid balance and electrolyte disturbance needs to be corrected. Renal disease/kidney disease/renal failure: kidneys fail to filter waste adequately from blood. Symptoms: Swelling, lack of urine production, fluid buildup; edema Treatment: Dialysis, kidney transplant. Renal Infarcts: The death of parts of the kidney due to lack of oxygen usually caused by a lack of blood supply. Symptoms: Nausea, vomiting, abdominal pain, flank pain, fever. Treatment: Support Management: dialysis. Renal Medullary Carcinoma: A tumor in the collecting duct of the nephron. Most common only in those with Sickle Cell Trait. Symptoms: 60% experience blood in the urine (hematuria), 50% have abdominal pain and 25% have significant weight loss. Treatments: When patients present a localized case, a surgical resection can be curative. Unfortunately, most cases are silent, and the cancer spreads past the point of curative care. Other Words you may hear: Hematocrit: A blood test that measures the amount of red blood cells in the blood, (% of whole blood made of red blood cells). Ischemia: Restriction of blood supply to the tissues. Ischemia of the kidney leads to necrosis, infarction. Isosthenuria: Non-concentrated urine, having the same osmolarity as plasma. Isosthenuria is usually a common sign of early renal failure. All information has been summarized from Medscape, the American Society of Hematology, and the Mayo Clinic. By Sean Marshall
This week the sickle cell anemia community had more international news to take in. Luckily there was good news that came fromlaboratoryequipment.com. The site’s news section mainly covers scientific and medical breakthroughs. Most news and academic article coverage is usually done by medical schools or professors. This generally means that the information is up to date for the time and is scientifically arcuate. The particular article was about the “two beneficial variants of a gene controlling red blood cell development.” The article explains that the newest study from King’s was all about the study of genomes of world populations. This was done to “look for the origin of changes in a key regulator gene that stimulates fetal haemoglobin production into adulthood.” To understand why there is even a study about fetal and adult haemoglobin the article explains what these two red blood cell types do. Fetal haemoglobin is the type of haemoglobin a human being produces in the womb. Hence the name fetal haemoglobin, however the average human body switches to producing adult hemoglobin after birth. It was explained in the article that “we continue to produce very small amounts of fetal haemoglobin in adulthood, some more than others. “ It also went on to explain how this ties into sickle cell anemia. “Patients who have the genetic factors that increase fetal haemoglobin production tend to have milder symptoms of their blood disorder.” This is an issue because according to the article “Patients who have the genetic factors that increase fetal haemoglobin production tend to have milder symptoms of their blood disorder.” Basically those who have blood disorders have bodies that are often producing more fetal hemoglobin then they can handle. This article is good news because it means the scientific community is narrowing down on potential ways to help people. In a quote from Stephen Menzal “"Patients who have milder versions of blood disorders, thanks to their ability to keep producing fetal haemoglobin, carry genetic clues that are helping us to understand the function of the genes and biological pathways involved in these diseases." It must be pointed out that the author of the article was King’s College, the very same place the study came from. This means that any negative impacts the study caused would not be reported on. This is good news but just remember to not take it all on face value alone. If you’d like to learn more about what was mentioned follow the link: http://www.laboratoryequipment.com/news/2014/07/beneficial-gene-has-spread-across-globe?qt-print_issue_laboutlook_supplemen=0 With the good news comes the bad. An article out of the Saudi Gazette explains that preliminary tests don’t stop genetic blood diseases. The Saudi Gazette first stated that “the latest statistics published by the Ministry of Health, the Saudi Ministry of Health, and the percentage of disease carriers is about 4.3 percent, and pre-marital screening tests have not stopped the spread of the disease.” It then has to be explained that The Saudi Ministry of Health launched “the mandatory premarital screening in 2004 in order to reduce the incidence of hereditary blood disorders.’ This covered diseases like sickle cell anemia and thalassemia. It also must be said that “in 2008, the program was expanded to include mandatory screening for hepatitis B and C viruses and Human Immunodeficiency Virus or more commonly known as HIV.” The article then got quotes from Internal medicine and infectious diseases consultant Dr. Dhiya Al-Hajjaj. She felt that “the lack of laws that prohibit the marriage of patients with sickle cell anemia and other serious hereditary diseases has contributed significantly to the increase in number of people infected with these diseases.” The article also said that Al-Hajjaj advised people who are infected with sickle cell anemia not to marry because of the potential negative impact on the next generation, especially since children born to couples carrying the disease have a 25 percent risk of infection. The bad news however isn’t that individuals with sickle cell anemia are on the rise or that people with blood disorders can be wed it’s the ideals behind the article. The solution to sickle cell isn’t a cure with homeopathic medicine or bone marrow transplants but officials trying to put laws into place forbidding people to marry, to have children because of a disease they are born with. The whole idea of destroying the disease is by stopping the blood lines of those who have it. This in bad news because this article reaches many people it gets the message that doctors and other experts feel that people with sickle cell should not wed one another. It’s not right and its dangerous thinking. If you would like to read the article for yourself follow to see if this is just one big overreaction go to the link below:http://www.saudigazette.com.sa/index.cfm?method=home.regcon&contentid=20140802213312 If you have any questions comments or concerns email us at [email protected] |
AboutThis section is solely to let our Sickle Soldiers tell their story trials & tribulations alongside things they feel are wrong in the Sickle Cell Community Archives
March 2016
Categories
All
|
 RSS Feed
RSS Feed
